


Critical Questions About Cassava's Recent Phase 2 Trial Readout: A Report
An In-depth Look at the Data and the Unanswered Questions That Remain

This report raises a series of concerns about Cassava Science’s trial readout and independent statistical analysis, some of which are based on simulated approximations of unreleased patient-level data. Some or all of these concerns may have ameliorative explanations. If this is the case, then I encourage the company to disclose, to the extent possible, trial data supporting any such explanations.
During the preparation of this report I contacted the biostatistician that performed the final trial analysis for Cassava Sciences, Dr. Suzanne Hendrix, but she was unable to comment on any of the trial data or statistical methods. She kindly offered to forward my inquiries to Cassava, but the company has yet to reach out.
Please note that the term deterioration (and sometimes decline, as found in the literature) in this report, indicative of a patient losing cognitive function, is associated with a positive change in score on the ADAS-Cog. Negative change in ADAS-Cog score is associated with improvement of cognitive function based on the test’s scoring system.
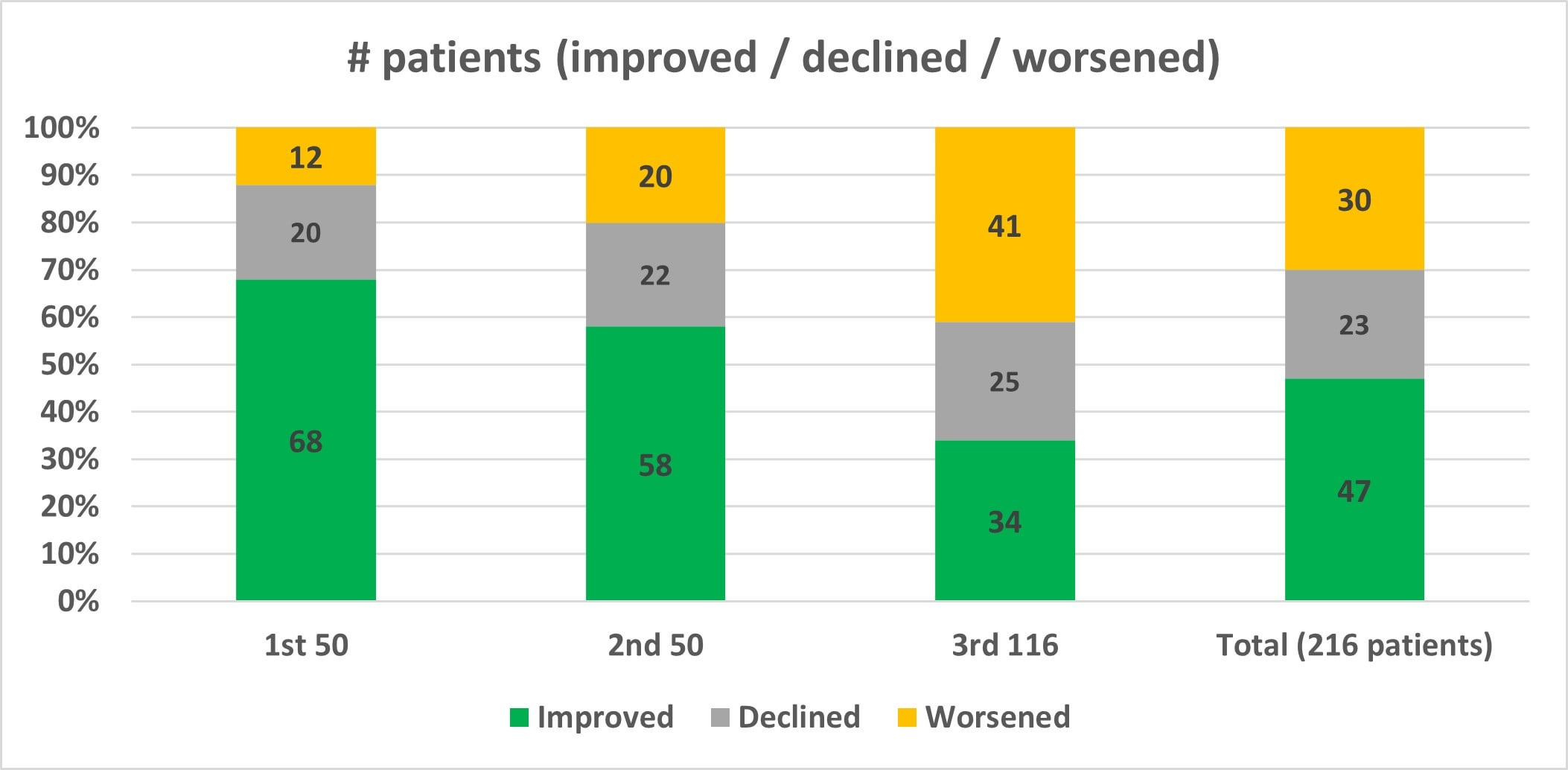
Cassava Sciences recently released the final topline readout from their Phase 2 open-label trial. In this final readout, ADAS-Cog change from baseline worsened for the second consecutive time since the release of the first 50-patient interim update. After the first 50 patients improved by an average of 3.2 points, and the next 50 deteriorated by an average of about 0.2-0.3 points (accounting for rounding uncertainty), we now see that apparently the final 116 patients deteriorated by an average of about 4.1-4.2 points, according to the data reported in Figure 1 of the final readout (which, somewhat confusingly and without explanation, does not align with the deterioration cited in the overall topline results). The worsening performance of the three groups is stark on its own, but it may actually be obscuring a precipitous decline in cognition for a small subset of patients that necessitates an explanation from the company. This potential safety issue is the most serious of a handful of concerns that I identified while scrutinizing the data from three press releases that Cassava issued about their Phase 2 trial of simufilam.
The report that follows is broken down into the following three parts:
Part I - Safety Concerns
Potentially significant anomalous deterioration in cognitive function among small group of patients.
Possible reporting issues and lack of clarity in Cassava’s external control data.
Part II - Trial Reporting Issues
Misleading endpoint characterization and trial reporting.
Unreported biomarker data and moving goalposts for primary endpoint.
Statistically significant unexplained variation in performance between all three trial cohorts.
Inappropriate post hoc subgroup analysis.
Potential misclassification of MCI as Mild AD.
Part III - Addressing Myths Propagating on Social Media
Misunderstanding of ADAS-Cog score distribution and probability of improvement on placebo.
Response analysis and identifying “responders” has no basis in reality.
Perils of relying on subgroup analyses, particularly a “mild AD” subgroup.
Part I: Uncovering Possible Safety Signal
Reverse Engineering the Patient Datasets
During Cassava’s H.C. Wainright conference talk, CEO Remi Barbier referenced “20% of patients really falling off a cliff” while appearing to temper expectations around the final topline readout for their Phase 2 of simufilam. If it is in fact the case that a much larger number of patients are experiencing a precipitous deterioration in cognitive function than would be expected, this could signal a serious risk for the patients currently participating in Cassava’s Phase 3 clinical trials. Generally speaking, any drug trial must account for the possibility that the chemical is actually making patients worse, not better—as Derek Lowe explains in an article discussing statistical analysis design, “the reality is that (1) you don’t know that your therapy might not make people worse, because (2) there have been many trials, in many therapeutic areas and through many different mechanisms, where that exact thing has happened.” In order to fully assess this risk, we will need access to patient-level data. However, Cassava has not provided this data, so instead we will need to do our best to approximate it from the various distribution summary statistics that Cassava reported in each of their three updates.
Disclaimer: What follows is exploratory in nature and should be interpreted with caution. Data found via optimization methods is not exact and is only an approximation of the underlying data.
To find approximate patient data, I wrote a stochastic optimization algorithm in Python. It’s essentially a directed random walk that will optimize under a set of conditions by incrementing/decrementing scores in pairs (holding the pre-specified mean constant) using the standard deviation to weight the transition probabilities. The objective is to find a dataset that fits the reported values—by definition, the real data can’t vary substantially from the values we get from the model without violating at least one of the reported quantities. I cannot guarantee that this method will find all valid datasets with equal probability, but in my evaluation the criteria that Cassava provided is specific enough, and the optimization scheme flexible enough, that optimization bias should be minimal. For the change from baseline in ADAS-Cog for the first 50 patients, I found the following approximate dataset (negative values represent improvement):
-13, -13, -13, -12, -12, -12, -11, -10, -10, -10, -10, -8, -8, -8, -7, -7, -7, -7, -6, -5, -5, -5, -4, -4, -4, -3, -3, -2, -2, -2, -2, -2, -2, -1, 0, 1, 2, 2, 2, 3, 3, 4, 4, 5, 6, 6, 7, 7, 9, 11This dataset compares to the one specified in the press release as follows:
Mean -3.16 (versus -3.2), standard deviation 6.34 (versus 6.3); 68% of values less than zero with mean -6.76 (versus -6.8) and standard deviation 3.81 (versus 3.8); and 20% between zero and 4 (both inclusive) with mean 2.5 for both and standard deviation 1.28 (versus 1.3).
For the first 100 patients, holding the first 50 constant at the above values and optimizing over the new 50, I found the next approximate set of 50:
-11, -11, -9, -8, -8, -7, -7, -6, -6, -5, -5, -5, -5, -4, -4, -4, -2, -2, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, 0, 1, 1, 2, 4, 4, 4, 4, 4, 4, 4, 5, 5, 6, 7, 9, 12, 13, 13, 14, 17Again, a comparison of the full dataset (this 50 and the prior 50) to the summary statistics in the press release:
Mean -1.45 (versus -1.5), standard deviation 6.62 (versus 6.6); 63% of values less than zero with mean -5.56 (versus -5.6) and standard deviation 3.77 (versus 3.8); and 21% between zero and 4 (both inclusive) with mean 2.71 (versus 2.7) and standard deviation 1.42 (versus 1.4).
Finally, for the last 116 patients Cassava presented the data in slightly different form, but we can convert the confidence interval (assuming it’s 95% CI and not 90% CI or another threshold—they didn’t specify) to a standard deviation of 9.64 and use the mean deterioration of 1.54 points to get a final approximate set of 116 that satisfies all known conditions:
-12, -11, -11, -10, -10, -8, -7, -5, -3, -3, -3, -3, -3, -3, -3, -3, -2, -2, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, -1, 0, 0, 0, 0, 1, 1, 1, 1, 2, 2, 2, 2, 2, 2, 3, 3, 3, 3, 3, 4, 4, 4, 4, 4, 4, 4, 4, 4, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 6, 16, 18, 22, 32, 38, 41, 47, 50, 63Potentially Significant Anomalous Deterioration in Cognition
The final six values (bolded in the prior dataset) represent a significant deterioration in cognition that warrants a closer look. Because Cassava was more specific in their reporting of the subgroup that improved their ADAS-Cog, along with the subgroup that deteriorated less than five points (i.e. scores between zero and four, inclusive), the population of patients declining in cognition by five points or more at week 52 will necessarily have greater variability under optimization. To help correct for the possibility that the above dataset was simply an outlier, I ran the final optimization step 100 separate times and averaged the results. Each time I started this step with the final 50 patients who scored 5 points or worse initialized at the same starting value (with a handful initialized a single point worse in order to maintain the correct total number of points, since it is not evenly divisible by 50). This initial condition should help reduce the variance within this final population and avoid the previously observed outliers as much as possible. Here are the ranked averages of this population:
5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.0, 5.01, 5.02, 5.09, 5.23, 5.88, 8.39, 13.66, 20.03, 24.72, 29.29, 34.39, 39.61, 45.92, 52.42, 63.34From the results it’s fairly clear that within these approximate datasets we have six patients extremely likely to have deteriorated by around 30 points or more on ADAS-Cog at 52 weeks. This result is highly alarming, as I will explain in more detail after establishing a suitable control distribution below. The inclusion criteria for this trial was mild-to-moderate Alzheimer’s disease with an MMSE score no less than 16, and the baseline ADAS-Cog distribution was comparatively mild (also covered in more detail below) at 19.1 ± 9.2 for a mild-to-moderate population. In order to determine the significance of these outliers, we first need to ascertain what the distribution parameters look like for changes in ADAS-Cog for mild-to-moderate Alzheimer’s disease patients after one year on placebo or without treatment. These datasets would be what are commonly referred to as “external controls.” I will circle back to the significance of this group of precipitously deteriorating patients after briefly covering issues related to Cassava’s selection and characterization of external controls, since the characteristics of a prototypical control group are essential for evaluating the significance of this deteriorative anomaly.
Issues in Cassava’s External Control Data
Obtaining a robust baseline placebo dataset to compare these values against is unfortunately not as straightforward as one would hope, and I could not verify any of the distribution summaries provided by Cassava’s consulting biostatistician. For example, in the Ito et. al. paper the results of the placebo decline in ADAS-Cog is indeed sourced from N = 19,972 patients as referenced in Cassava’s press release, but the estimated decline and accompanying confidence intervals appear to be obtained via 100 simulated datasets, not directly from the underlying data. The confidence interval reported, assumed to be 95% CI (this was again not specified), of (3.99, 5.16) with N = 19,972 would suggest a standard deviation of 42.2, rendering this dataset essentially meaningless on a test that is bounded between 0 and 70. Based on the literature, where standard deviations of ADAS-Cog scores tend to be between 5 and 10, it seems unlikely that the reported values are correct.
The solanezumab EXPEDITION 1 and EXPEDITION 2 trial analysis reported ADAS-Cog changes at week 80, not week 52, and the supplementary material with a week 52 value does not appear to specify whether error bars are SE, SD, 95% CI, or 90% CI, and values do not appear to align with what is reported in Cassava’s press release (I could very well have missed some data, but I looked pretty hard for it). It is not clear how Cassava obtained their estimate or whether they imputed patients, since the N that Cassava reported for each trial aligns with the starting population, while only 73.1% and 77.1% of placebo patients finished EXPEDITION 1 and 2, respectively. It is possible that an interpolative adjustment was made by the consulting biostatistician, however this is not specified anywhere in Cassava’s press release and requires clarification. In general, the press release is scant of any details about which models or statistical methods were used, when these method were applied, under what assumptions, and with what choices for free parameters.
The study by Thomas et. al. on longitudinal decline is behind a paywall, and I was unable to locate any detailed ADAS-Cog data for Cortexyme’s (now Quince) failed atuzaginstat trial.
It is actually remarkably difficult to find a straightforward external 52-week measurement for an ADAS-Cog11 (other variants generally contain a different number of questions, making direct comparison challenging) placebo comparator with the proper baseline MMSE and full summary statistics to compare with Cassava’s data, which on its own undermines the meaningfulness of this open-label, non-placebo-controlled trial for which Cassava is relying solely on their choice of external controls. The best that we can do is make an inference from trials or observational studies that approximate the conditions, but fall short of direct comparison in at least one critical way.
Cognition Deterioration Anomaly Analysis Using Suitable External Control Distributions
For one reasonable comparator, we can look at the aducanumab placebo data. This data is from a milder population according to initial MMSE scores, but it is also measured at week 78 as opposed to week 52, which offsets this difference to some degree. The reported mean deterioration in ADAS-Cog13 with associated standard deviation for each trial is 5.16 ± 9.4 and 5.14 ± 8.9, which is fairly generous to Cassava given that the placebo deterioration cited across all data in their press release is less than five. In a 2019 position statement co-authored by Dr. Hendrix—the biostatistician who performed the final trial analysis—it is posited that "the rate of decline on the ADAS-cog has changed over the past 2 decades from 7 points per year to 4.5 points per year, although the standard deviation has remained constant at 6-7 points." I should also note that ADAS-Cog13 scores on a scale from 0 to 85, which may inflate the expected decline we see above and partly account for the worse than expected deterioration.
Looking back at the six patients that likely deteriorated by about 30 points or more, the associated probability of obtaining a change in ADAS-Cog greater than 30 for the aducanumab placebo distributions under a Gaussian model is approximately 0.4% and 0.3%, respectively; for a change greater than 40, it is 0.01% and 0.005%. A Bernoulli trial of 216 tests with 6 success at probability 0.004 is p < 0.0003, and at probability 0.003 is p < 0.0001. And a trial of 216 tests with 3 succeeding at probability 0.0001 is p < 0.00001. This strongly suggests that these six patients were extremely unlikely to have deteriorated more than 30 points over one year simply by chance.
If we assume that, for example, the Thomas et. al. 2016 ADCS study distribution is accurate as presented, it would imply a placebo distribution of 3.89 ± 7.2, which correlates to a less than 0.02% chance of obtaining a value greater than 30 for any single observation. The probability of obtaining six in 216 tries would be vanishingly small.
These patients that are “falling off a cliff,” as CEO Barbier insinuated, raise serious concerns about the effects of simufilam, and a closer audit of the patient-level data is warranted.

Part II: Misleading Trial Characterization and Analysis
Improper Endpoint Characterization and Incomplete Reporting
In their press release, Cassava characterized the trial as “an open-label safety study with exploratory efficacy endpoints.” This is a mischaracterization of the goals of this Phase 2 trial based on both the common definition of a Phase 2 trial, and on Cassava’s own trial description. According to the FDA framework, a Phase 2 trial is meant to establish “efficacy and side effects.” While the side effects speak to the safety endpoint, efficacy at this stage is not considered merely “exploratory.” This is echoed by, for example, the University of Cincinnati College of Medicine in their description of trial phases, where they explain that “Phase II studies determine the effectiveness of an experimental drug on a particular disease or condition in approximately 100 to 300 volunteers.”
Cassava aligned with these authorities when they designed their trial—the detailed description of the trial begins with a statement that “the objectives of this study are to build the safety database for simufilam (PTI-125) and to investigate its effects on biomarkers, cognition and neuropsychiatric symptoms” [emphasis added]. To wit, one of the primary endpoints is listed as “change from baseline in ADAS-Cog-11 during open-label period 1.” Referring to the FDA guidance on how to treat multiple endpoints in clinical trials, it states that “when there are many endpoints prespecified in a clinical trial, they are usually classified into three families: primary, secondary, and exploratory.” Far from an exploratory endpoint, change in cognition from baseline is a primary outcome measure in this trial. As such, it requires a full statistical characterization by the sponsor prior to any claim of trial success (including the much more equivocal claim by Cassava of “positive results”).
Further, most of the trial analysis was focused on ADAS-Cog, with only a short paragraph devoted to safety, belying the notion that the cognition endpoint was simply exploratory and not a critical part of the claim that the results of the trial were “positive.” Despite the amount of energy devoted to characterizing changes in ADAS-Cog, Cassava avoids making any concrete statements about whether or not this endpoint produced a statistically significant result. Granted, the trial design, with its lack of a placebo group and no pre-specified comparators, would make any such claim suspect, but even still Cassava presents data that quite clearly is not significantly different from its own choice of external comparators. Looking at Figure 1, the confidence interval for simufilam does not clear two of the intervals among comparators (though one of them is the aforementioned Ito 2016 group that is likely erroneous), and barely clears two others. There are ample issues with essentially unbounded multiplicity (i.e. the comparator group can apparently be any placebo group of the sponsor’s choosing) in the trial design, and yet even with boundless degrees of freedom the final result does not achieve statistical significance across almost half of the reported groups. For any reasonable claim of success, it would need to clear all of them, or have a clearly defined pre-specified statistical analysis plan that integrates them into a single comparator group, which the trial does not.
The company should clarify on what specific basis they consider the results “positive” with regards to a statistically significant result in the ADAS-Cog primary endpoint.
Unreported Biomarker Endpoints, Moving Goalposts
When Cassava first registered its Phase 2 trial in May 2020, it had only a single primary endpoint, “Cerebrospinal fluid P-tau, neurofilament light chain, neurogranin, Total Tau, YKL-40, Abeta42 (pg/mL).” Safety and ADAS-Cog were added approximately six months later (again belying the claim that the only non-exploratory aspect of this trial was safety). In June 2021, this endpoint was updated to end at month 6 for all patients, and a rider was tacked onto the endpoint specifying that it would only be measured in the first 25 patients. The modification of this endpoint after more than a year—and four trial description updates—raises serious questions about whether it can be trusted. Yet, Cassava continues to use this small subgroup in order to claim success on a primary endpoint, even going so far as to include p-values of p < 0.00001 despite measuring barely more than 10% of the trial population. Further, this subset of 25 patients apparently improved nearly 5 points, strongly discordant with the overall deterioration of 1.5 points in the full population. All biomarker data have also been removed from Cassava’s corporate presentation (as have ADAS-Cog data), where they were featured prominently for the last year and a half. An explanation for their removal and for the change in the endpoint is warranted.
Looking again at the detailed trial description, we see the unqualified statement that “plasma biomarkers will be evaluated in all subjects.” This evaluation is missing. Also in the trial description is the claim that “the first 50 subjects will also provide a CSF sample at Month 6 or Month 12 for evaluation of change from baseline in CSF biomarkers.” These data are incomplete.
Most secondary endpoints, such as those relating to SavaDx, are omitted from the trial analysis entirely. While these are less important than the primary endpoints, they are still listed in the official trial documentation and should be explicitly accounted for by the company, even if they were simply abandoned.
Significant Unexplained Variation Between Trial Cohorts
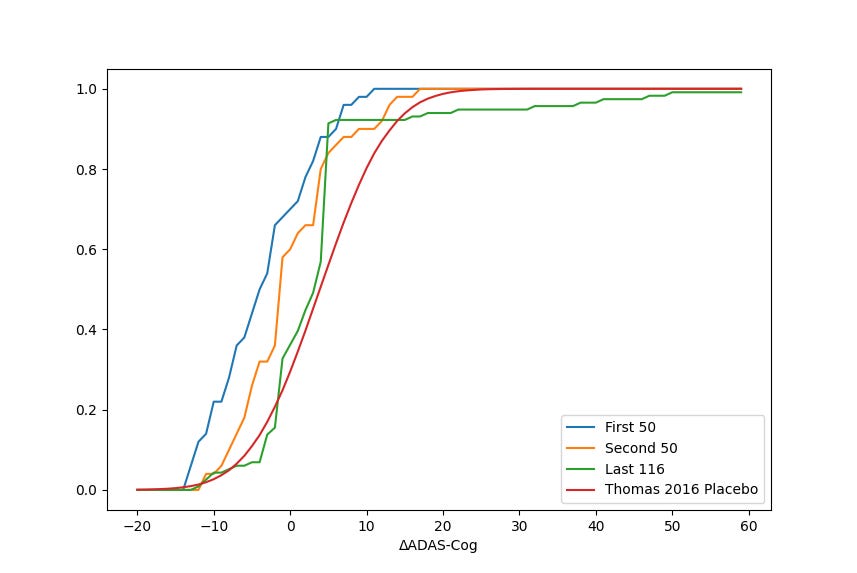
As discussed earlier, each of the three cohorts deteriorated more, on average, than the cohort that preceded it. In a vacuum, declining performance across three data releases may not in and of itself be cause for concern. Given the backdrop of federal investigations which started after the first update—and assumedly continued ramping up as more patients finished the trial—it’s worth understanding how different the three cohorts appear in order to assess the possibility that trial population monitoring was overly lax prior to the involvement of federal regulatory bodies.
Using a standard two-tailed t-test for difference in the mean of the candidate populations ascertained above, we find that cohort 1 and 2 have a different underlying mean with p < 0.01, cohort 2 and 3 with p < 0.006, and cohort 1 and 3 with p < 0.001, implying that each of these three sets of patients differed in a statistically significant way from the other two. We can visualize these changes by plotting the cumulative distribution function (CDF) for the three trial cohorts, along with the CDF for the Thomas 2016 placebo distribution for reference. The variation of the final cohort in particular is notable, as it tends to follow the placebo curve for improved or stable scores until the significant deviation for the rapidly declining patients noted above. Kolmogorov-Smirnov tests for these three cohorts using their CDFs return similar p-values for statistically significant difference, at p < 0.02, p < 0.02, and p < 0.0001 for the three cohort comparisons. Additional details about patient selection and screening should be provided in order to reconcile these significant variations.
Inappropriate Post Hoc Subgroup Analysis
In an apparent attempt to bolster the trial results, Cassava performed a subgroup analysis—shown in Figure 2—which separated mild patients post hoc and compared the change in their ADAS-Cog score to a different set of external control groups from those used in the full analysis. As an exploratory exercise, there is nothing inherently wrong with identifying subgroups—it is fairly routine, as I’ll discuss later, for sponsors to identify promising subgroups and design new trials around them. However, subgroup analysis is fraught with well-documented issues that compromise its meaningfulness when not properly accounted for. First, subgroups should be pre-specified for analysis in order to control for multiplicity. Post hoc subgroup selection is noted in Wang et. al. as “of particular concern because it is often unclear how many were undertaken and whether some were motivated by inspection of the data.” This alludes not only to issues of multiplicity, but to the ability of the sponsor to choose subgroup parameters after unblinded inspection of the data.
In this case, Cassava used MMSE score to identify subgroups, but did not pre-specify the ranges for mild versus moderate until after the completion of the trial (nor was the subgroup analysis itself pre-specified—they could just as well have stratified by APOE carriers, age, biomarkers, etc. in order to “datamine” a positive result). Cassava chose MMSE 21-26, however this is far from a unique range within the literature or even Cassava’s own comparator trials. For example, J.L. Cummings, who previously served on Cassava’s Scientific Advisory Board before its abrupt dissolution, defines mild-to-moderate Alzheimer’s disease as falling within MMSE 12-24. In the ENGAGE trial cited as a comparator, inclusion criteria included MMSE 24-30. The gantenerumab Phase 3 trial comparator has MMSE ≥ 22 as an inclusion criteria. These trials also frequently used additional inclusion criteria to ensure that the target population fell within the correct disease stage, such as clinical dementia rating-global score (CDR-GS) of 0.5 or 1.0 in the gantenerumab trial. The recently completed lecanemab Phase 3 trial included “CDR score of 0.5 to 1.0 and a CDR Memory Box score of 0.5 or greater” and “NIA-AA core clinical criteria for probable Alzheimer's disease” among inclusion criteria, along with MMSE 24-30. Even Cassava’s own Phase 3 trial defines a different MMSE range for inclusion, this time 16-27, stretching one point “milder” than the mild subgroup here. These differences are not merely academic, either—without showing us the MMSE scores that correlate with each ADAS-Cog outcome, we can’t rule out Cassava having chosen the range strategically after inspecting the data. Remember, ADAS-Cog is highly variable, and the more degrees of freedom in the analysis the more likely that we’re simply digging up artifacts of the noise inherent to ADAS-Cog evaluation.
For additional context about why post hoc subgroup analyses are not appropriate determinants of trial success, it is worth recounting the retrospective on aducanumab by Knopman et. al. that speaks directly to a doomed “mild vs. moderate” post hoc analysis in the EXPEDITION trials. From the text:
Post hoc analyses are not equivalent in their predictive power to primary pre-specified analyses. As participants in different arms of a trial often differ in one or more relevant variables—such as age, sex, cognitive severity, baseline Aβ PET levels, APOE genotype, biological phenotype, or any interactions between these variables—claims based on a post hoc contrast within one of these variables must be considered untrustworthy.
Recent experience with solanezumab should be a cautionary reminder of the fickleness of claims based on post hoc analyses. After a pair of phase III trials failed to achieve their goals, review of the unblinded data suggested that mild, but not more advanced, patients benefited from solanezumab. These observations spawned a third trial, EXPEDITION 3, that was restricted to mild AD dementia. Unfortunately, it too failed to meet its goals.
Finally, subgroup analysis should be driven by identification of a biologically relevant factor for differentiation, and Cassava made no such attempt to buttress their analysis of the mild subgroup with any biologically motivated analysis. The claimed method of action for simufilam involves “stabilizing a critical protein in the brain,” but Cassava provided no reason to expect the stabilization of a protein to aid in recovery for mild Alzheimer’s patients and not moderate ones. There is no “attenuating factor” for protein stabilization in the mild disease stage beyond the oft-cited “catching the cascade early” hypothesis, and this hypothesis has yet to be verified in any clinical trial.
Cassava’s subgroup analysis violates a host of best practices. The mild subgroup was chosen post hoc and was not a pre-specified endpoint or stratification variable; it has no biologically motivated differentiation factor external to the metric itself; and no appropriate controls for multiplicity or unblinding were included. Further, Cassava’s subsequent Phase 3 trial does not plan for such a subgroup analysis (MMSE is a randomization stratification variable, but not an endpoint stratification) and is not designed for FDA approval based on performance within any particular subgroup. If the mild subgroup is meant to establish the success of the trial, Cassava should explain how and why. Otherwise, this analysis is moot.
Potential Misclassification of MCI as Mild AD
In reporting the subgroup of patients that fell into the “mild” category Alzheimer’s disease, Cassava states that the starting ADAS-Cog score for this group was 15 ± 6.3. Perhaps due in part to lax screening criteria, this ADAS-Cog distribution appears to be more closely aligned with mild cognitive impairment (MCI) than other comparative studies of mild Alzheimer’s disease. One study, for example, found the ADAS-Cog11 scores of mild AD patients to be 19.66 ± 6.3, almost 5 points worse on average than Cassava’s mild subgroup. For the mild subgroups in solanezumab’s EXPEDITION 2 trial, the ADAS-Cog distribution summary statistics for the placebo and treatment arms were 19 ± 7 and 20 ± 7, respectively. In a more general 2015 review paper that modeled correlation between MMSE, ADAS-Cog, and CDR-SOB, the results have an ADAS-Cog score of 15 correlated to an MMSE of 26, which means that the average patient in this cohort according to ADAS-Cog fell on the mildest edge of the specified MMSE window. Based on the mild cohort’s summary statistics, it remains within the realm of possibility that a number of the patients in this cohort would barely qualify as cognitively impaired according to their ADAS-Cog score.
We know from the Ito 2016 paper, cited numerous times by Cassava, that cognitive decline tends to accelerate as AD progresses, meaning that patients starting with worse ADAS-Cog baseline scores tend to also lose more points in subsequent testing. The large discrepancy between baseline ADAS-Cog scores in Cassava’s mild subgroup, and potentially in their trial group overall, needs to be accounted for.

Part III: Addressing Myths Propagating on Social Media
This section, in contrast to the previous two, does not pertain to issues that Cassava could reasonably be expected to address directly. Cassava is not, generally speaking, responsible for incorrect or poorly conceived analyses by its shareholders or other members of the public. That said, the lack of clarity and misleading characterizations of the trial covered to this point encourage the kind of shambolic analysis addressed in this section.
Misunderstanding of Variability in ADAS-Cog Change Over Time
In the presentation of their trial results, Cassava has consistently broken down the population into three subsets—those that improved their ADAS-Cog scores, those that deteriorated by less than five points, and those that deteriorated by five points or more. This has led some Cassava enthusiasts to undertake analyses that compare the percentage of patients that improved to the percentage that improve in other trials. First of all, this comparison is not an appropriate measure of the success or failure of the drug. Probability of improvement on ADAS-Cog is not an endpoint in either the Phase 2 trial or the ongoing Phase 3 trial of simufilam, and, to my knowledge, has never been a primary endpoint in any serious Phase 2 or Phase 3 trial in Alzheimer’s disease. The correct outcome measure is the change in the mean score of the entire population. The analysis of mean change in the population essentially supersedes whatever analysis one might consider performing for a binary outcome (e.g. logistic regression) in the sense that it already contains all of the useful information about improvement probability encoded into the mean improvement. For example, imagine a population where 70% of patients improve, but the population declines by the same amount as placebo on average. This would imply that the 30% of patients that deteriorated did so at a much more rapid pace than expected. Perhaps this will encourage identification of a hidden confounding factor, but it will certainly not help make the drug approvable in and of itself. A new series of trials would be required, and would be undertaken only when the confounding factor is identified.
Beyond this misunderstanding of what measures are actually pertinent to the efficacy evaluation of the drug, there is also general misinformation circulating regarding the percentage of patients that might be expected to improve on ADAS-Cog at one year without treatment. ADAS-Cog is a very noisy test, and given the 4.5 ± 7 guideline placebo change at one year provided by Dr. Hendrix earlier, we would expect 26% of patients to actually improve, and just over 50% to deteriorate by less than five points. In recent trials the percentages of improvers have at times likely been much higher based on the reported summary statistics in those trials. Yes, Alzheimer’s is an irreversible degenerative disease. No, this does not correlate to monotonic decline on ADAS-Cog, because again, it is a highly variable test. This point is even emphasized in an FDA position statement co-authored by Dr. Hendrix:
Another study detected a total of 108 errors were made by 80.6% of the 72 raters and concluded that most experienced raters made at least one error that may affect ADAS-cog scores and clinical trials outcomes.
I should also note something of importance regarding an open-label trial with ADAS-Cog as a primary endpoint—it is subject to significant scoring bias pressure. Meaning, evaluators know that every patient they are evaluating is taking the drug, and they score their evaluations with this knowledge. This can have a wide range of possible influence on the scoring outcomes, but the main point is that it is another significant source of uncontrolled variance that decreases the reliability of the results.
There Is No Basis for Identifying “Responders”
Extending the logic above, it should hopefully be clear that responder analysis is a futile exercise for this trial. There will always be “responders” when evaluating patients using ADAS-Cog, but identifying these responders is almost always an exercise in classifying noise. This problem only gets worse the more mild the population, as ADAS-Cog becomes less reliable with less severe cognitive impairment. One need only look at the quantile regression performed on the solanezumab trial data to get a sense of how futile this exercise really is—despite improvement maintained at 80 weeks for the top 30th percentile of mild AD patients, nobody is under the illusion that solanezumab will be approved anytime soon. These are not “responders,” they are noise artifacts.
The Perils of Subgroups and a Tiresome Pivot to Mild AD
As yet another cautionary tale encompassing a number of issues reflected in this report, consider the development and eventual abandonment of azeliragon. Early development showed promise, but the high dosage arm was dropped due to safety signals and faster than expected cognitive deterioration. The lower dose showed no benefit at the conclusion of the trial, but a signal showed up upon further evaluation of the mild AD subgroup. The drug received fast-track designation, but it failed to meet its endpoints in a Phase 3 trial focused on mild AD patients. Then, investigators identified another subgroup—type 2 diabetes and elevated concentrations of acetylated hemoglobin—which showed a significant effect and had the potential to modulate the drug target (i.e. biological motivation). A new Phase 2/3 trial was run, and again it missed its endpoints. Development of azeliragon was scrapped.
A comprehensive review by Kwon et. al. helps elucidate why subgroup analyses, like the two spawned by azeliragon, so often fall flat despite appearing so enticing:
With some AD drugs, reliance on post hoc subgroup analyses of failed trial data without further verification of the results led to the initiation of phase III trials that ultimately failed. It is common practice to scrutinize negative trial results to detect treatment responsive subgroups and other insights for the design of future trials. However, there is substantial risk of spurious results with such analyses, as subgroups have not undergone the same rigors of recruitment and randomization as the original groups, are subject to smaller sample sizes, and multiple statistical comparisons violate the assumptions implicit in statistical tests, increasing the likelihood of spurious “significant” results.
Or, we could look directly at what Dr. Hendrix has to say about post hoc subgroups in a contributing chapter of Alzheimer's Disease Drug Development:
Often, subgroup analyses are used to try to salvage a mostly negative study, but this approach is usually futile since a negative subgroup can often be found that counteracts the positive subgroup that is promoted.
Another example provided in the Kwon 2022 review is tarenflurbil, for which the Phase 3 trial was modified to include only mild AD patients after a post hoc analysis of the Phase 2 data, but it still failed to meet its endpoints and development has since been discontinued. The review goes on to note that ADAS-Cog may be a questionable choice of endpoint for mild or prodromal Alzheimer’s disease, stating that “as AD trials have targeted patients earlier in the disease progression, the sensitivity of [ADAS-Cog] has been questioned for milder affected patients.” Perhaps this is why we see such a defined pattern of pharmaceutical companies pivoting to mild disease after their drug fails to make an impact overall. Yet another example of this is the recent failure of troriluzole, for which Biohaven claimed that there were “potentially promising results among just the patients with mild disease” only to remove the drug’s Alzheimer’s indication from their pipeline shortly thereafter. Or crenezumab, which recently failed a Phase 3 trial that was initiated after “phase II results did not show a significant benefit in treatment compared to placebo but a post hoc analysis showed a benefit in mild AD subjects with an MMSE range of 22–26,” as described in a Mehta et. al. review paper. The review, like most others, flags issues with ADAS-Cog variability and unreliability, devoting an entire section to “Problems with Raters” in ADAS-Cog testing.
The ill-fated post hoc subgroup analysis (and subsequent trial flop) is something of a rite of passage for Alzheimer’s drugs, likely in no small part because of the inherent variance in the most important outcome measures related to cognitive function. When, for example, TauRx hopped first from mild-to-moderate AD to mild AD and then to patients not taking AChEIs in a series of trials leading up to CTAD 2016, scientists at CTAD had seen enough—Paul Aisen, Editor-in-Chief of CTAD sponsor The Journal of Prevention of Alzheimer’s Disease, felt strongly that the subgroup analyses were inappropriate, telling Alzforum that “the reported subgroup analyses are, in fact, uninformative, because they lacked an appropriate placebo group.” Dr. Hendrix was similarly critical of TauRx’s claims when speaking with Alzforum, which prompts the question—what specifically did Dr. Hendrix identify in Cassava’s data that permits a mild subgroup analysis using a small sample size with no placebo control arm, in a population dubiously mild for AD, and with an apparently subpar moderate group counterbalancing the mild one? To adapt a wryly fitting quote, “If there is evidence of a treatment effect […] to support moving into another Phase 3 study, either overall or in a subgroup, it has not been presented publicly.”
Given your real reasoned concern for the five patients with ADAS-Cog scores that fell off the cliff, are you surprised that Phase 3 trials passed their interim Data and Safety Monitoring Board reviews?